
GLM vs. t-tests vs. non-parametric tests if all we care about is NHST
A skeleton simulation of different strategies for NHST for count data if all we care about is a p-value, as in bench biology where p-values are used to simply give one confidence that something didn’t go terribly wrong (similar to doing experiments in triplicate – it’s not the effect size that matters only “we have experimental evidence of a replicable effect”).
tl;dr - At least for Type I error at small \(n\), log(response) and Wilcoxan have the best performance over the simulation space. T-test is a bit conservative. Welch is even more conservative. glm-nb is too liberal.
load libraries
library(ggplot2)
library(ggpubr)
library(MASS)
library(data.table)
library(cowplot)The simulation
- Single factor with two levels and a count (negative binomial) response.
- theta (shape parameter) set to 0.5, 1, 100
- Relative effect sizes of 0%, 50%, 100%, and 200%
- Ref count of 4, 10, 100
- \(n\) of 5, 10, 20, 40
p-values computed from
- t-test on raw response
- Welch t-test on raw response
- t-test on log transformed response
- Wilcoxan test
- glm with negative binomial family and log-link
do_sim <- function(niter=1, return_object=NULL){
# the function was run with n=1000 and the data saved. on subsequent runs
# the data are loaded from a file
# the function creates three different objects to return, the object
# return is specified by "return_object" = NULL, plot_data1, plot_data2
methods <- c("t", "Welch", "log", "Wilcoxan", "nb")
p_table_part <- matrix(NA, nrow=niter, ncol=length(methods))
colnames(p_table_part) <- methods
p_table <- data.table(NULL)
res_table <- data.table(NULL)
beta_0_list <- c(4, 10, 100) # control count
theta_list <- c(0.5, 1, 100) # dispersion
effect_list <- c(1:3, 5) # relative effect size will be 0%, 50%, 100%, 200%
n_list <- c(5, 10, 20, 40) # sample size
n_rows <- length(beta_0_list)*length(theta_list)*length(effect_list)*length(n_list)*niter
sim_space <- expand.grid(theta_list, beta_0_list, effect_list, n_list)
plot_data1 <- data.table(NULL)
plot_data2 <- data.table(NULL)
debug_table <- data.table(matrix(NA, nrow=niter, ncol=2))
setnames(debug_table, old=colnames(debug_table), new=c("seed","model"))
debug_table[, seed:=as.integer(seed)]
debug_table[, model:=as.character(model)]
i <- 0
for(theta_i in theta_list){
for(beta_0 in beta_0_list){
# first get plots of distributions given parameters
y <- rnegbin(n=10^4, mu=beta_0, theta=theta_i)
x_i <- seq(min(y), max(y), by=1)
prob_x_i <- dnbinom(x_i, size=theta_i, mu=beta_0)
plot_data1 <- rbind(plot_data1, data.table(
theta=theta_i,
mu=beta_0,
x=x_i,
prob_x=prob_x_i
))
# the simulation
for(effect in effect_list){
for(n in n_list){
beta_1 <- (effect-1)*beta_0/2 # 0% 50% 100%
do_manual <- FALSE
if(do_manual==TRUE){
theta_i <- res_table[row, theta]
beta_0 <- res_table[row, beta_0]
beta_1 <- res_table[row, beta_1]
n <- res_table[row, n]
}
beta <- c(beta_0, beta_1)
treatment <- rep(c("Cn", "Trt"), each=n)
X <- model.matrix(~treatment)
mu <- (X%*%beta)[,1]
fd <- data.table(treatment=treatment, y=NA)
for(iter in 1:niter){
i <- i+1
set.seed(i)
fd[, y:=rnegbin(n=n*2, mu=mu, theta=theta_i)]
fd[, log_yp1:=log10(y+1)]
p.t <- t.test(y~treatment, data=fd, var.equal=TRUE)$p.value
p.welch <- t.test(y~treatment, data=fd, var.equal=FALSE)$p.value
p.log <- t.test(log_yp1~treatment, data=fd, var.equal=TRUE)$p.value
p.wilcox <- wilcox.test(y~treatment, data=fd, exact=FALSE)$p.value
fit <- glm.nb(y~treatment, data=fd)
debug_table[iter, seed:=i]
debug_table[iter, model:="glm.nb"]
#if(fit$th.warn == "iteration limit reached"){
if(!is.null(fit$th.warn)){
fit <- glm(y~treatment, data=fd, family=poisson)
debug_table[iter, model:="poisson"]
}
p.nb <- coef(summary(fit))["treatmentTrt", "Pr(>|z|)"]
p_table_part[iter,] <- c(p.t, p.welch, p.log, p.wilcox, p.nb)
}
p_table <- rbind(p_table, data.table(p_table_part, debug_table))
p_sum <- apply(p_table_part, 2, function(x) length(which(x <= 0.05))/niter)
res_table <- rbind(res_table, data.table(beta_0=beta_0,
beta_1=beta_1,
n=n,
theta=theta_i,
t(p_sum)))
} # n
} # effect
plot_data2 <- rbind(plot_data2, data.table(
theta=theta_i,
mu=beta_0,
n_i=n,
beta1=beta_1,
x=treatment,
y=fd[, y]
))
}
}
if(is.null(return_object)){return(res_table)}else{
if(return_object=="plot_data1"){return(plot_data1)}
if(return_object=="plot_data2"){return(plot_data2)}
}
}
do_it <- FALSE # if FALSE the results are available as a file
if(do_it==TRUE){
res_table <- do_sim(niter=1000)
write.table(res_table, "../output/glm-t-wilcoxon.txt", row.names = FALSE, quote=FALSE)
}else{
plot_data <- do_sim(niter=1, return_object="plot_data2")
res_table <- fread("../output/glm-t-wilcoxon.txt")
res_table[, n:=factor(n)]
}
#res_tableDistribution of the response for the 3 x 3 simulation space
# extreme inelegance
mu_levels <- unique(plot_data[, mu])
theta_levels <- unique(plot_data[, theta])
show_function <- FALSE
show_violin <- TRUE
if(show_function==TRUE){
gg1 <- qplot(x=x, y=prob_x, data=plot_data[mu==mu_levels[1] & theta==theta_levels[1],], geom="line")
gg2 <- qplot(x=x, y=prob_x, data=plot_data[mu==mu_levels[2] & theta==theta_levels[1],], geom="line")
gg3 <- qplot(x=x, y=prob_x, data=plot_data[mu==mu_levels[3] & theta==theta_levels[1],], geom="line")
gg4 <- qplot(x=x, y=prob_x, data=plot_data[mu==mu_levels[1] & theta==theta_levels[2],], geom="line")
gg5 <- qplot(x=x, y=prob_x, data=plot_data[mu==mu_levels[2] & theta==theta_levels[2],], geom="line")
gg6 <- qplot(x=x, y=prob_x, data=plot_data[mu==mu_levels[3] & theta==theta_levels[2],], geom="line")
gg7 <- qplot(x=x, y=prob_x, data=plot_data[mu==mu_levels[1] & theta==theta_levels[3],], geom="line")
gg8 <- qplot(x=x, y=prob_x, data=plot_data[mu==mu_levels[2] & theta==theta_levels[3],], geom="line")
gg9 <- qplot(x=x, y=prob_x, data=plot_data[mu==mu_levels[3] & theta==theta_levels[3],], geom="line")
}
if(show_violin==TRUE){
gg1 <- ggviolin(x="x", y="y", data=plot_data[mu==mu_levels[1] & theta==theta_levels[1],], add="jitter")
gg2 <- ggviolin(x="x", y="y", data=plot_data[mu==mu_levels[2] & theta==theta_levels[1],], add="jitter")
gg3 <- ggviolin(x="x", y="y", data=plot_data[mu==mu_levels[3] & theta==theta_levels[1],], add="jitter")
gg4 <- ggviolin(x="x", y="y", data=plot_data[mu==mu_levels[1] & theta==theta_levels[2],], add="jitter")
gg5 <- ggviolin(x="x", y="y", data=plot_data[mu==mu_levels[2] & theta==theta_levels[2],], add="jitter")
gg6 <- ggviolin(x="x", y="y", data=plot_data[mu==mu_levels[3] & theta==theta_levels[2],], add="jitter")
gg7 <- ggviolin(x="x", y="y", data=plot_data[mu==mu_levels[1] & theta==theta_levels[3],], add="jitter")
gg8 <- ggviolin(x="x", y="y", data=plot_data[mu==mu_levels[2] & theta==theta_levels[3],], add="jitter")
gg9 <- ggviolin(x="x", y="y", data=plot_data[mu==mu_levels[3] & theta==theta_levels[3],], add="jitter")
}
gg_example <- plot_grid(gg1, gg2, gg3, gg4, gg5, gg6, gg7, gg8, gg9,
nrow=3,
labels=c(paste0("mu=", mu_levels[1], "; theta=", theta_levels[1]),
paste0("mu=", mu_levels[2], "; theta=", theta_levels[1]),
paste0("mu=", mu_levels[3], "; theta=", theta_levels[1]),
paste0("mu=", mu_levels[1], "; theta=", theta_levels[2]),
paste0("mu=", mu_levels[2], "; theta=", theta_levels[2]),
paste0("mu=", mu_levels[3], "; theta=", theta_levels[2]),
paste0("mu=", mu_levels[1], "; theta=", theta_levels[3]),
paste0("mu=", mu_levels[2], "; theta=", theta_levels[3]),
paste0("mu=", mu_levels[3], "; theta=", theta_levels[3])),
label_size = 10, label_x=0.1)
gg_example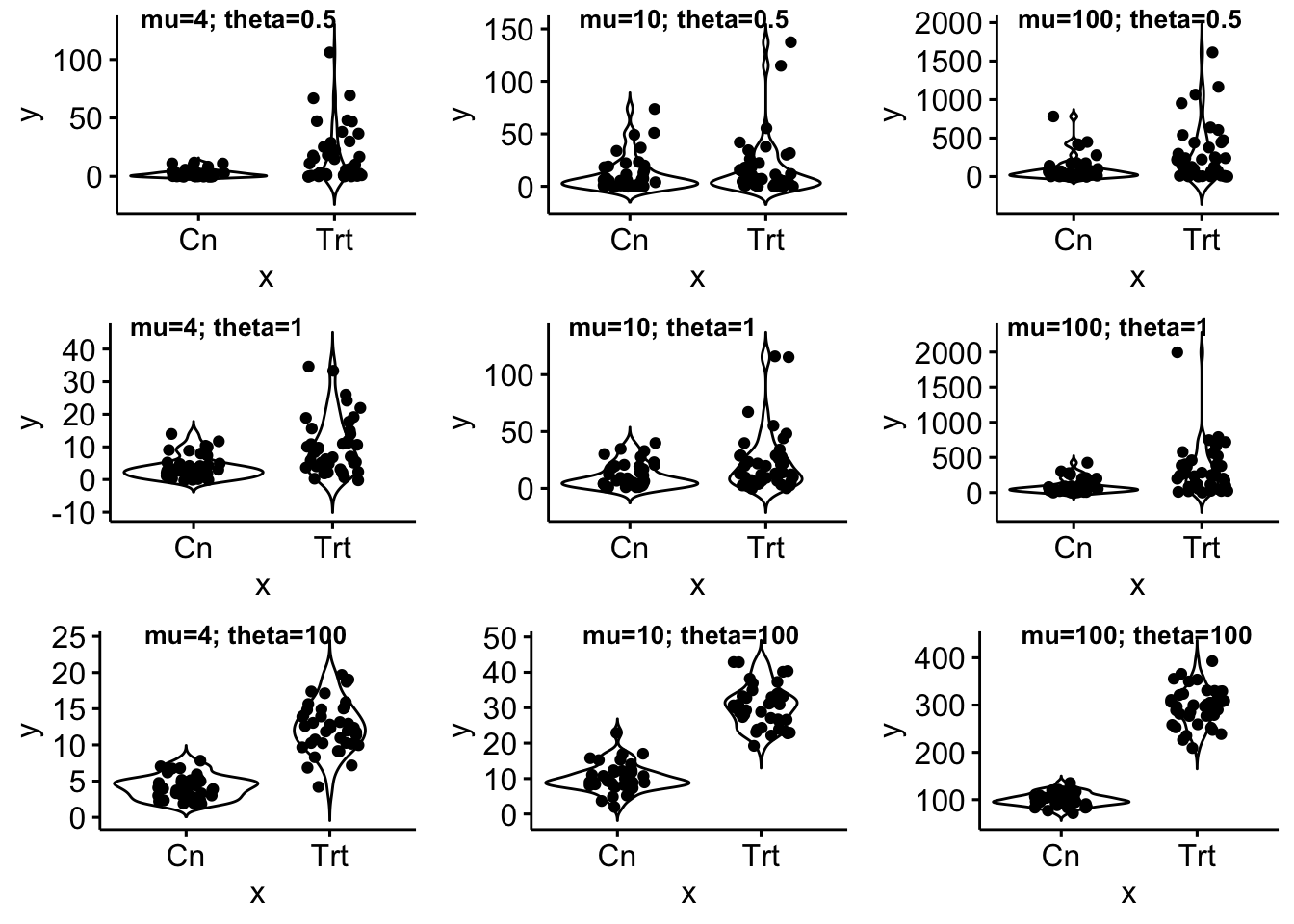
Type I error
res <- melt(res_table,
id.vars=c("beta_0", "beta_1", "n", "theta"),
measure.vars=c("t", "Welch", "log", "Wilcoxan", "nb"),
variable.name="model",
value.name="frequency")
# res[, beta_0:=factor(beta_0)]
# res[, beta_1:=factor(beta_1)]
# res[, theta:=factor(theta)]
# res[, n:=factor(n)]gg <- ggplot(data=res[beta_1==0], aes(x=n, y=frequency, group=model, color=model)) +
geom_line() +
facet_grid(beta_0 ~ theta, labeller=label_both) +
NULL
gg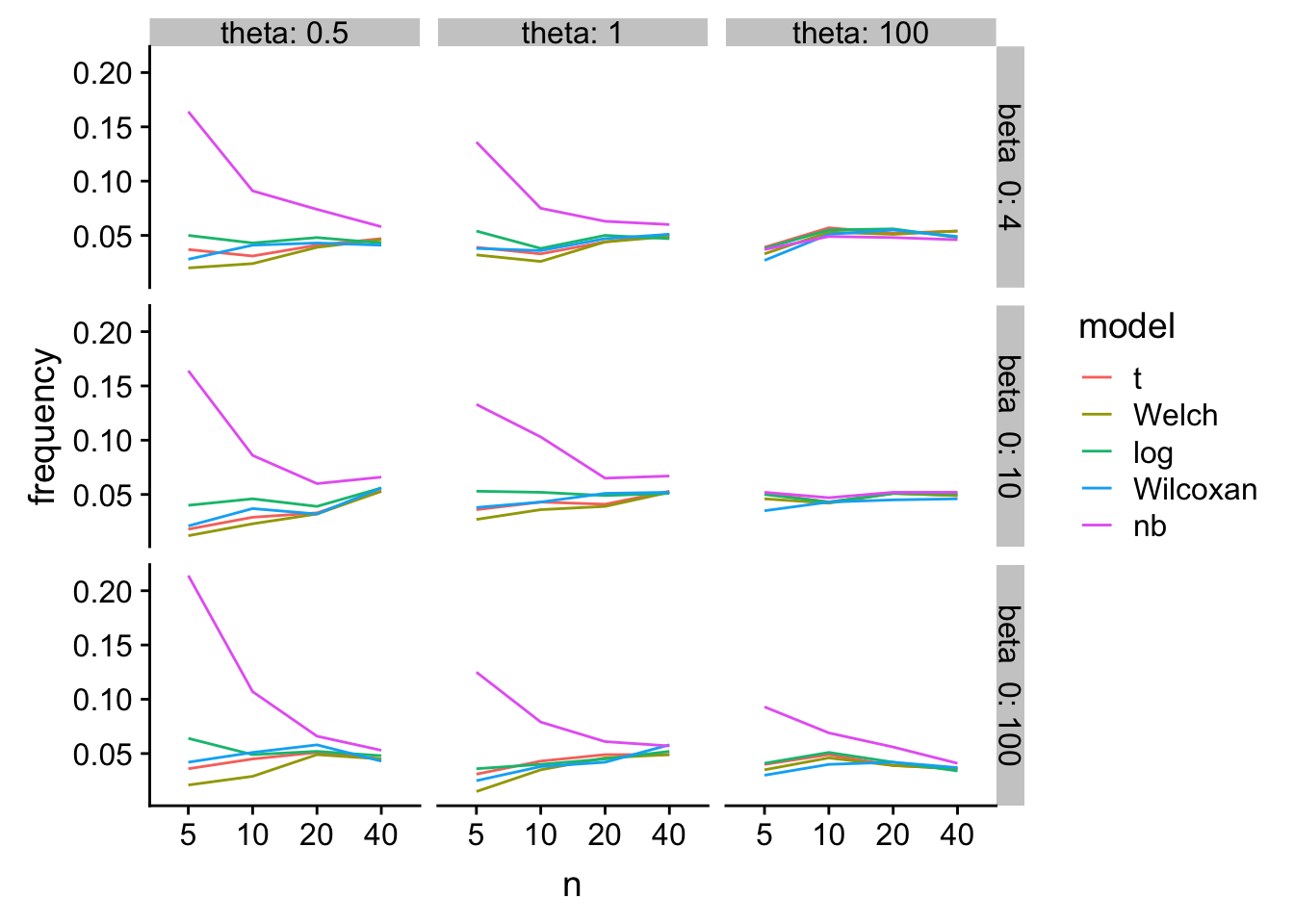
Ouch. glm-nb with hih error rates especially when n is small and the scale parameter is small
Power
b0_levels <- unique(res$beta_0)
# small count
gg1 <- ggplot(data=res[beta_0==b0_levels[1] & beta_1 > 0], aes(x=n, y=frequency, group=model, color=model)) +
geom_line() +
facet_grid(beta_1 ~ theta, labeller=label_both) +
NULL
# large count
gg2 <- ggplot(data=res[beta_0==b0_levels[3] & beta_1 > 0], aes(x=n, y=frequency, group=model, color=model)) +
geom_line() +
facet_grid(beta_1 ~ theta, labeller=label_both) +
NULL
gg1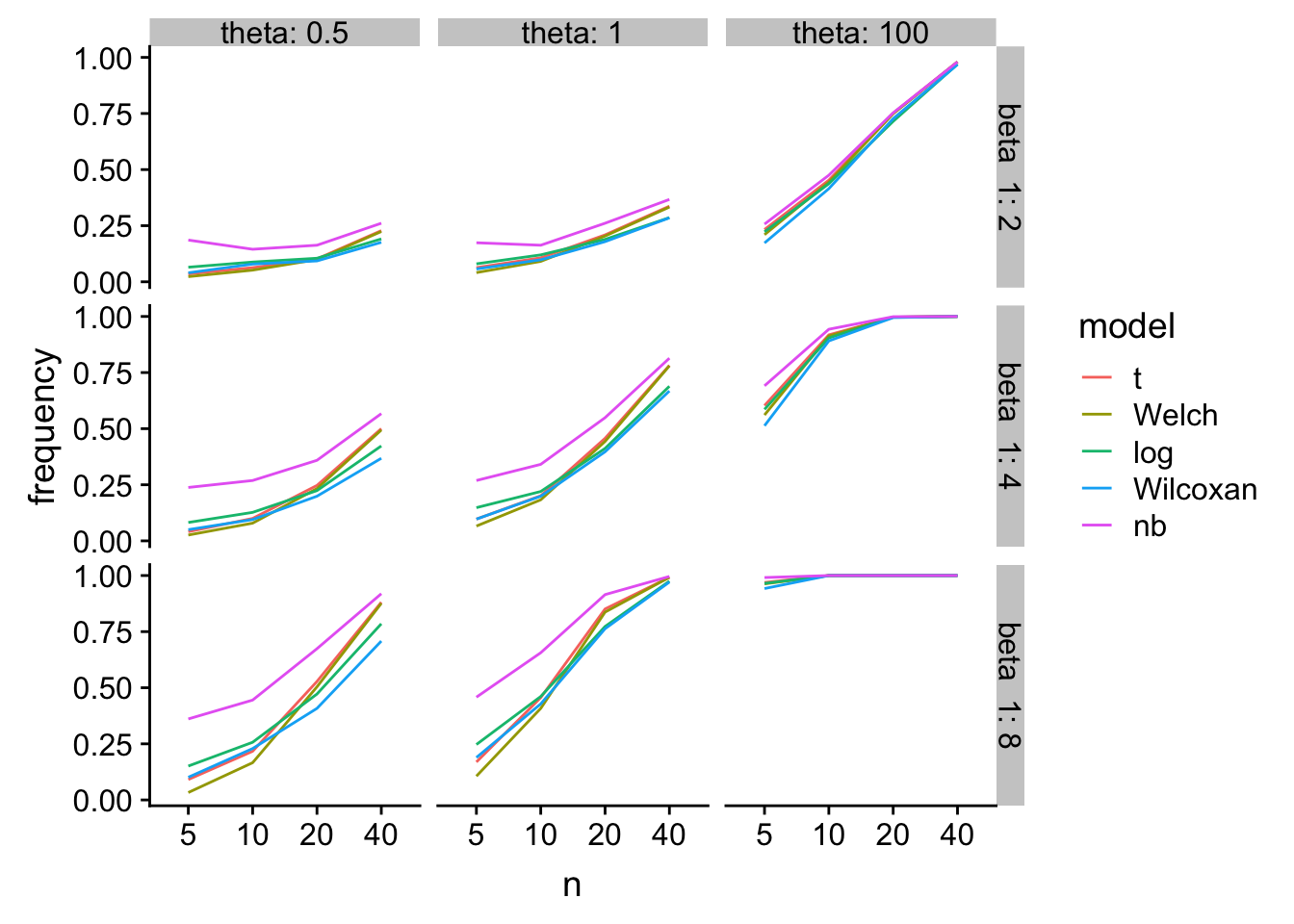
gg2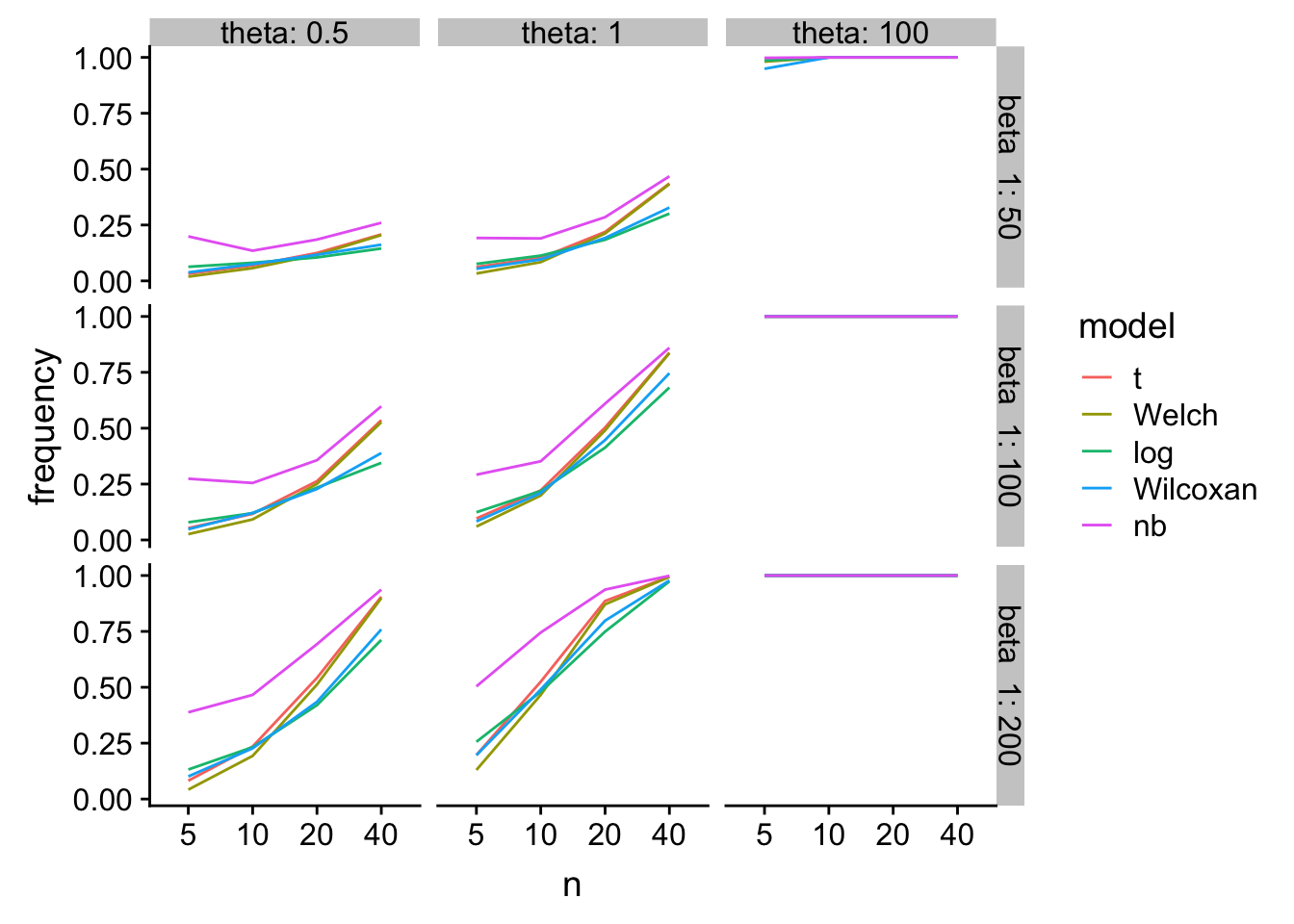
glm-nb has higher power, especially at small n, but at a type I cost.
